-
-
- Evolution balances the benefits of virulence in parasites (greater rate of shedding) against the disadvantages (shedding for less time, and less dispersal)
- “Short-sighted selection” increases virulence. “Far-sighted selection” increases transmission.
- Viruses that jump to new hosts are sometimes extraordinarily virulent. Well-established viruses clearly moderate their pathogenicity.
- The benefits to the virus of moderation will increase as we approach herd immunity.
- Milder CoV-2 strains that are more temperature-sensitive may arise spontaneously in the next few weeks or months.
-
One idea that I haven’t heard mentioned much in the last few weeks is the virulence-transmission trade-off model of selection. This describes the evolution of pathogenicity that we should expect in parasites, including viruses. It’s clear that strains that replicate themselves very quickly will cause the host to emit more (progeny) viruses early on in the illness. It’s also likely that, on average, fast-growing strains will cause worse illnesses. However, such aggressive strains are likely to cause illness that immobilizes their hosts, so they may, paradoxically, be spread less widely. The model suggests that the benefits (to the virus, not to us) of greater virulence – more rapid progression of the illness and “shedding” of viruses in greater numbers – need to be balanced against the disadvantages [see scientific reference 1, below].
This is largely about timing: if transmission is very fast compared to the development of the illness – because contacts between susceptible hosts are frequent (think, for example, of chickens in a barn) – selection becomes a race between the different strains to become infectious as quickly as possible after transmission. If, on the other hand, the virus has relatively few chances to jump to susceptible new hosts (think wandering albatrosses), milder strains will be favored so that shedding can carry on for longer. This extended shedding increases the chance that the host will still be infectious when they meet another potential host. In real-life chicken barns, very virulent strains of bird flu and other illnesses can develop very quickly, resulting the the death of almost every bird in a few days.
So if we behave more like chickens in a barn, we expect the virus to become more virulent. If we’re more like wandering albatrosses, less virulent.

Barn interior showing the suffocation by foam of chickens infected with avian influenza. Picture by Roee Shpernik.
Note that the selection towards reduced virulence is not normally driven by the death of hosts – that takes too long. Rather, it’s about how long hosts remain infectious – because we believe that slow-growing strains also tend to be less virulent. A recent study [ref. 15] into CoV-2 shedding found that the length of shedding was indeed related to the severity of the disease: the median period of CoV-2 shedding by asymptomatic patients was 19 days, whereas for symptomatic patients it was 14 days.
There’s another way to look at this: the viruses within each host are, in evolutionary terms, competing with each other. Selection within the host tends to favor more virulent sequences, because they tend to replicate faster. This is referred to as “short-sighted” selection, because it works well (from the point of view of the competing viruses) in the short term. In order to survive, however, viruses also need to transmit themselves to other hosts. Here, less aggressive strains are favored, resulting in “far-sighted” selection. The final result is a trade-off between short-sighted and far-sighted evolution. That may be why coronavirus has a “proofreading” mechanism, which reduces copying errors and so prevents too much short-sighted selection.
I created a simple model of CoV-2 infection, which can be found here: https://oldwivesandvirologists.blog/a-simple-model-of-cov2-transmission/
This view seems to be correct, because when viruses first appear in a new host – after jumping from one species to another – they are quite often extraordinarily virulent. Ebola, SARS, myxomatosis in European rabbits, Lassa hemorrhagic fever, and bird flu are all examples of this. In comparison to these illnesses, most well-established “endemic” viruses are mild. It seems to take time for milder strains to appear, so that an equilibrium can be reached in the new host species. If all viruses were as virulent as Ebola, multicellular animals, from earthworms to mammals, might well be impossible. Over time, milder strains have come to the fore, and we have ended up with viruses similar to the 100 (largely unrelated) strains that cause the common cold. They are active enough to make us sick, but they don’t usually kill us.
The point about reducing the mobility of hosts might be particularly important in humans. For example, we are probably less likely to get on an airplane and travel to the far side of the world with a very active strain that makes us sick within a few hours, than with one that brews slowly and allows us to walk around shedding viruses for several days – simply because the incubation period of the milder illness is longer. The transported, milder, strain might then start a new epidemic in a remote location. If this process is repeated many times and on different scales, you can imagine that we will end up with intermediate strains – neither very passive nor very virulent. An equilibrium is reached. Nowadays, at least in richer societies, we have the luxury of going to bed when we get a fever, rather than being forced to work (which was often the case in the past and is still the case in some poorer communities). This might also be expected to reduce virulence over time.
According to this view, the extreme virulence of Spanish influenza in 1918 was partly the result of the selective conditions that existed at that time. People tended to “soldier on” when they had fevers, because they were determined to contribute to the war effort.
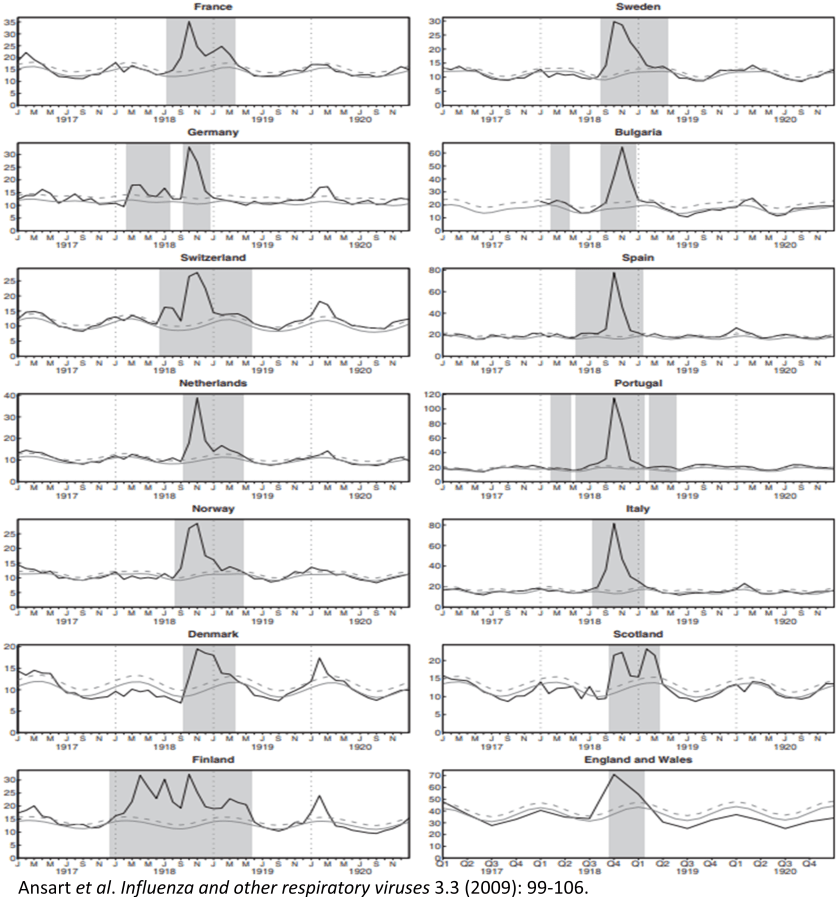
Mortality in 12 European countries, 1917-1920.
An example of the rapid transmission of Spanish influenza was reported in the British Medical Journal in 1918 [2] by two doctors who shared a train compartment from London to York with a sick airman, who said he had flu. Two days later both doctors had bouts of flu, and two days after that, the wife and two children of one of the doctors were also sick with the same illness. That particular strain of influenza was very unusually virulent and fast-acting, and may have been subject to unusual selective pressures – resulting in a dramatic increase in virulence and mortality. Hope-Simpson [3, 4] noted in the 1970s that influenza infections in institutions tend to be unusually severe. This puzzled him, but it may be that when residents live and sleep close together, selection favors virulent strains that develop fast, because they can be transmitted whether or not they cause severe illness. Examples might include military barracks, crowded refugee camps, boarding schools, care homes and hospitals. No matter how sick the patients become, the virus particles that they shed may be able to reach other nearby residents. This is rather like the chicken barn with bird flu, where every host becomes infected. We should also worry about hospitals, which can become the human equivalent of chicken barns – epidemics there can easily become very dangerous [1]. In the 1918 Spanish flu epidemic, it was reported that patients treated in an “open-air” (tented) hospital had a much higher survival rate than those in conventional hospitals [5].
There are other considerations here. Although the evidence has been ignored by most virologists, for many decades scientists have noted that many “wild” strains (as opposed to most laboratory-bred strains) of respiratory viruses are temperature-sensitive [e.g. refs 6-8; coronaviruses were first isolated at 33°C, ref. 9. For many other references see my paper, ref. 10.]. That’s to say, they’re more active at lower temperatures (typically 33 – 35°C) than at body temperature (around 37°C). This makes sense because they live in the nose and throat, which are some of the coldest parts of the body. The virus needs to stay out of the heart and lungs, so that the host will continue to move around, in order to maximize its transmission, as discussed above. Temperature-sensitivity is, potentially, a very good way to make sure that this happens. In fact there is one good bit of evidence that almost all respiratory viruses use temperature in this way: the simple observation that almost all respiratory viruses are more common in winter than summer. This makes sense intuitively, because if they’re more active at low temperatures, then, as ambient temperature falls in the autumn, epidemics become more likely. Conversely, in spring, when the temperature rises, all temperature-sensitive viruses will become less active, and cold epidemics rarer. Note that respiratory viruses have a high mutation rate, and we can expect them to adjust their pathogenicity quickly to suit their local climate and environment. A 2011 scientific review acknowledged that there was no satisfactory scientific explanation for the seasonality of influenza [11]. After that article appeared, I published a review of seasonality in respiratory viruses in Medical Hypotheses, which focused on the natural temperature-sensitivity of respiratory viruses [10]. My blog article gives a more informal description for the layperson [12]. Seasonality is clearly important, because if we can find the degree of temperature-sensitivity of Covid 19, we may be able to predict the course of the epidemic when the weather warms up in the summer – and, even more importantly, when the temperature drops in the autumn.
If some strains of SARS-CoV-2 develop greater temperature-sensitivity several things may happen: (i) they may be more likely to colonize the nose and throat, because these are relatively cold parts of the body, rather than the lungs. This can increase sneezing and runny noses, increasing transmission; (ii) they may cause a mild infection that the host doesn’t notice, allowing him or her to go to work, or get on a train or plane while already shedding virus; and (iii) they may be less likely to give rise to dangerous “systemic” infections where the virus spreads into the internal organs of the body.
Note for the technically-minded: a lot of scientific attention has focused in the last few weeks on the sequences of the proteins that the virus makes. However, like influenza, coronaviruses have untranslated regions of RNA with secondary structure that is conserved [13]. This means that it must be doing something. Secondary structure is inherently temperature-sensitive, and many organisms (both microorganisms and higher organisms) make use of ‘‘RNA thermometers” [14]. These are RNA segments that respond to temperature changes with three-dimensional conformational changes that alter gene expression. Because many individual bases contribute to each conserved RNA configuration, the virus has the potential to fine-tune its temperature-sensitivity by changing bases in these regions. These regions might be good places to look for mutations that give rise to milder strains. We should also look at sub-optimal codon and dinucleotide usage by the virus.
These ideas have practical implications for dealing with the current epidemic. They suggest that milder strains of CoV-2 will arise spontaneously and, in principle, their selection can be encouraged. In the next few weeks we may hear a lot more in the media and from scientists about less virulent CoV-2 strains. A recent report from Zhejiang University found that the most aggressive strains of Sars-CoV-2 generated 270 times as much viral load as the least potent type. The CoV-2 strains that are present in, say, Germany may be milder than those in Italy, simply because the authorities have more efficiently picked up the most obvious cases, allowing a greater proportion of mild strains with long incubation periods to slip through the net. We should bear in mind that the various CoV-2 strains are competing with each other. Whichever arrives in a particular host first will begin to generate immunity, which will reduce the chance of infection by other strains. If mild strains can spread, herd immunity will increase, increasing the advantage of milder strains that give shedding for longer periods. If we focus on tracking, tracing and isolating individuals carrying the most aggressive strains that cause serious illness, we may be able to “flip” the trade-off equilibrium towards low pathogenicity and so speed up the arrival of milder strains. Once milder strains establish themselves it’s less likely that virulent ones would be favored by natural selection (although the fall and winter may offer opportunities for more virulent strains).
All this needs to be taken into account in designing measures to reduce mortality in the current epidemic. In the long-run it opens up new opportunities to understand viral respiratory illnesses at a fundamental level.
Patrick Shaw Stewart, Sunday 15 March 2020. (Edited 31 April).
Scientific references –
[1] Frank, Steven A. “Models of parasite virulence.” The Quarterly review of biology 71.1 (1996): 37-78.
[2] Macdonald, Peter, and J. C. Lyth. “Incubation period of influenza.” British medical journal 2.3018 (1918): 488.
[3] Hope-Simpson, Robert Edgar. “Epidemic mechanisms of type A influenza.” Epidemiology & Infection 83.1 (1979): 11-26.
[4] Hope-Simpson, R. E. “Age and secular distributions of virus-proven influenza patients in successive epidemics 1961–1976 in Cirencester: epidemiological significance discussed.” Epidemiology & Infection 92.3 (1984): 303-336.
[5] Hobday, Richard A., and John W. Cason. “The open-air treatment of pandemic influenza.” American journal of public health 99.S2 (2009): S236-S242.
[6] Richman, Douglas D., and Brian R. Murphy. “The association of the temperature-sensitive phenotype with viral attenuation in animals and humans: implications for the development and use of live virus vaccines.” Reviews of infectious diseases 1.3 (1979): 413-433.
[7] Stern, H., and K. C. Tippett. “Primary Isolation of Influenza Viruses at 33° C.” Lancet (1963): 1301-2.
[8] Kung, H. C., et al. “Influenza in China in 1977: recurrence of influenzavirus A subtype H1N1.” Bulletin of the World Health Organization 56.6 (1978): 913.
[9] Tyrrell, D. A. J., and Parsons, R. “Some virus isolations from common colds. III. Cytopathic effects in tissue cultures.” Lancet (1960): 239-42.
[10] Shaw Stewart, Patrick D. “Seasonality and selective trends in viral acute respiratory tract infections.” Medical hypotheses 86 (2016): 104-119.
[11] Tamerius, James, et al. “Global influenza seasonality: reconciling patterns across temperate and tropical regions.” Environmental health perspectives 119.4 (2011): 439-445.
[12] https://oldwivesandvirologists.blog/
[13] Yang, Dong, and Julian L. Leibowitz. “The structure and functions of coronavirus genomic 3′ and 5′ ends.” Virus research 206 (2015): 120-133.
[14] Narberhaus, Franz, Torsten Waldminghaus, and Saheli Chowdhury. “RNA thermometers.” FEMS microbiology reviews 30.1 (2006): 3-16.
[15] Long, Quan-Xin, et al. “Clinical and immunological assessment of asymptomatic SARS-CoV-2 infections.” Nature Medicine (2020): 1-5.
Click to access s41591-020-0965-6.pdf
______________________________________
_____________________________________
The Hypothesis
For a general discussion of the seasonality of respiratory viruses, written for the layperson, please see
Every winter, colds and flu increase
For detailed scientific information about the seasonality of respiratory viruses, including discussion of the trade-off model, viral dormancy and much else, see my 2016 paper:
For a discussion of the strange timing and duration of influenza epidemics, please see
The strange arrivals – and departures – of influenza epidemics in the UK, 1946-1974
Applications to Covid-19
For information about the probable seasonality of Covid-19, and whether we can expect it to become rarer in the summer, or reappear in the fall, please see
Predicting the seasonality of Covid-19
For comments about the epidemiology of Covid and other respiratory illnesses, please see
Epidemiology of respiratory illness
For discussion of how the trade-off model can be applied to the Covid epidemic see
Covid 19 and the trade-off model
For a simple model of the transmission of viruses such as CoV-2, please see
A simple model of CoV-2 transmission
For comments about how quickly we can expect viruses to adapt to new environments, please see
Adaptability or respiratory viruses
For more detailed scientific points about CoV-2, see
Technical notes on CoV-2 for scientists
For practical tips on avoiding respiratory illness see
Suggestions for avoiding colds and flu – and Covid-19

One thought on “Covid 19 and the trade-off model of selection”