By Patrick Shaw Stewart, October 2022 – February 2023.
Part 1, preface: the evolution of high-fidelity replication (first draft)
I wrote this section last, and it applies to both higher organisms (Part 2, below) and quasispecies-forming entities such as viruses (Part 3). I suggest that high-fidelity genetic replication is a very valuable resource for all organisms and viruses and easily lost. They therefore put a lot of energy into conserving high fidelity, and re-acquiring it by recombination when it is lost. However, in this section I want to tackle a fundamental question, which I have not seen discussed in the literature: how can natural selection ever increase the fidelity of replication ? I ask this because selective pressures and genetic drift would be expected to degrade fidelity at times, so it must be possible for natural selection to counteract this tendency. To understand the difficulty, consider the following thought-experiment: imagine a virus with its own polymerase complex, having an error-rate of 1-incorrect-nuclotide-in-5-billion. Call it strain A. It mutates and gives rise to strain B, which has an error-rate of 1-in-4-billion. These two strains are at first identical in every other way. Then strain A mutates again and gives rise to strain C, with an error-rate of 1-in-6 billion. After a few rounds of replication, strain A progeny have on average 5 mutations, strain B 6 mutations, and strain C, 4 mutations. The difference in the number of mutations is small, and the system is noisy, with some mutations having much more effect (positive or negative) than others. How can natural selection (over time) consistently favour A over B, and C over the other two? I’m going to try to answer this. My answer is in two parts: (1) I suggest that high fidelity can only evolve during periods of relative evolutionary “stasis”, when a biological species is already very well-adapted to its environment, and very few mutations are beneficial (i.e. almost all are harmful). (2) I also suggest that repeated serial founder effects can favour high-fidelity. Figure 1.1 illustrates the first point: when strong selective pressures exist, a higher-than-usual proportion of (all) mutations are likely to be beneficial. A novel lower-fidelity polymerase complex will provide more mutations, so the strain likely to be selected at first (indicated by the red lineage Figure 1.1A). However, as mutations accumulate in essential genes, the fitness of such a lineage declines. Moreover, the high mutation rate likely gives rise to mutations the polymerase genes themselves, decreasing fidelity still further, eventually resulting in an “error catastrophe”. In my example, the red lineage becomes extinct. Meanwhile lineages with only a few mutator mutations (purple) may survive. Contrast this with the situation for a well-adapted species (Figure 1.1B). Since almost all mutations are now harmful, the lower-fidelity strains (green) rapidly become extinct. In this case, I suggest that polymerase mutations that increase fidelity are more likely to be selected (blue lineage).
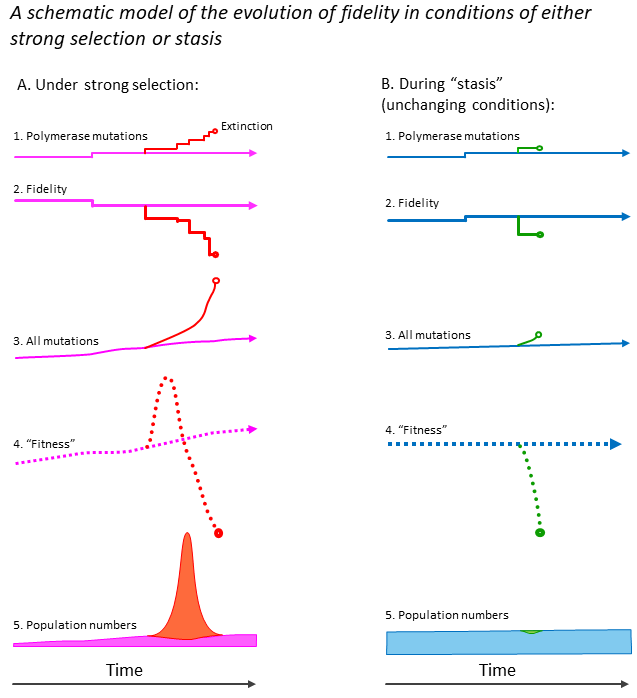
Figure 1.1. A schematic model of the evolution of fidelity, contrasting conditions that encourage strong genetic selection, with unchanging conditions that provide evolutionary stasis. The model suggests that stasis favors the evolution of high-fidelity genetic replication. Consider first the effect of strong selection as an organism adapts to new conditions (e.g. an animal or plant that colonizes an island), or a virus as it adapts a new host (panels A1-A6). If an important polymerase mutates to a significantly more error-prone form (shown in the red lineage of panels A1 and A2), further mutations are expected to appear in the polymerase gene itself. This is expected to result in a rapid increase in mutation throughout the genome (panel A3). During rapid adaptation a higher proportion of mutations is expected to be beneficial. Since a rapidly-mutating lineage can adapt more rapidly, its “fitness” increases at first (panel A4, red lineage). However, further mutations in essential functions will decrease fitness at later times, resulting in the extinction of lineages with very high mutation rates. The population of a such a lineage may increase rapidly, but then collapse (panel A6, red lineage). (As an aside, the beneficial mutations may be “rescued” by recombination between low (red) and high-fidelity (purple) lineages, as discussed in the main text.) In other cases, biological entities may be well-adapted to their environment or host (examples would be animals that live in environments that are stable over geological time-scales, or viruses that infect one stable species for millennia). Here the model suggests a different situation, known as stasis, which is shown in panels B1-B6. Almost all mutations are now harmful, therefore any mutation conferring low fidelity (panels B1 and B2, green lineage) may provide an almost instant selective disadvantage, acting after one or a few mutations. Since such an environment is likely to be highly competitive, the low-fidelity strain quickly becomes extinct. The model therefore suggests that occasional mutations that confer increased fidelity can be selected, such that high fidelity can evolve. Note that I refer above to mutations in polymerases for simplicity, but mutations in other associated proteins such as proof-reading endonucleases may similarly reduce fidelity. Panel B2 shows a polymerase mutation that increases fidelity in the green lineage. This is not intended to suggest that such mutations are more common during stasis – indeed the opposite is likely. However, the model suggests that any mutations increasing fidelity that do arise are more likely to be conserved during stasis.
Figure 1.2 shows how serial genetic bottlenecks might increase fidelity. Low-fidelity replication may be neutral or beneficial in the short-term, but disastrous in the medium-to-long-term. The regular foundation of new “colonies” therefore increases the time interval during which selection operates (even though there are more red arrows, the blue arrows are longer and give rise to new colonies more often). It follows that the frequent foundation of new colonies can promote the selection of higher-fidelity replication.
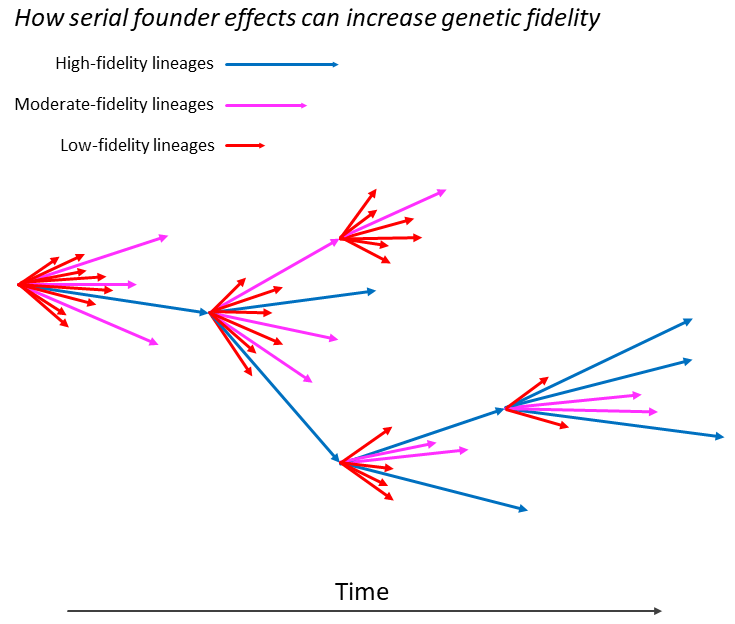
Figure 1.2. A model showing how serial founder effects can encourage the evolution of increased genetic fidelity. The figure illustrates how a series of genetic bottlenecks, occurring as a biological entity (an organism or virus) passes from one community or host to another, can increase fidelity. At earlier times (left-hand side) more low-fidelity lineages are present. At later times (right-hand side), after a series of transfers to new communities or hosts, more high-fidelity lineages are present after selection. There are two reasons why higher-fidelity lineages may be more likely to found new colonies: (1) higher-fidelity lineages may survive longer since low-fidelity lineages may suffer “error catastrophes” and become extinct; (2) low-fidelity lineages may adapt more rapidly to their local community or host, but, in the process (via so-called “antagonistic pleiotropy”), they may lose the functions that are required to jump to new communities or hosts. Examples of animals experiencing serial founder effects include those that undergo annual migrations such as arctic terns, salmon and monarch butterflies. Viruses may experience serial founder effects when virions are transferred from host to host. The model suggests that, in some instances, low-fidelity virions do not retain the functionality to survive the transfer and establish successful infections.
Note: I am aware that polymerase mutations that increase fidelity but slow up replication sometimes appear and have been selected in laboratory experiments. For example, Fitzsimmons and colleagues found that polioviruses with the 3DG64S mutation in the polymerase had higher fidelity but replicated much more slowly than the corresponding wild-type. I regard this as a special case where fidelity increased at a cost, and the mechanism of replication has not fundamentally improved. I suggest that it is unlikely that increased fidelity could evolve by accident in a laboratory setting, especially when viruses are propagated in cells that come from animals that are not their natural hosts. It would be interesting to deliberately select for high fidelity – somehow!
Part 2: The Preservation of High-Fidelity Replication in Animals and Plants (First Draft)
Key insight
The more genes are involved in the mechanism that transfers genetic material between individuals – including genes that usually do something else – the lower the chance that defective DNA polymerase genes with mutator mutations will be transferred.
Abstract
Mutations must occasionally arise in all genes, including the genes that encode the proteins that replicate genetic material – the nucleic acid polymerases and their associated proteins. Some of these mutations must reduce the fidelity of DNA replication (or RNA replication in RNA viruses), without actually being fatal. Lineages that replicate with reduced fidelity must therefore exist in all biological kingdoms, although we do not know how common they are. A fundamental problem at the heart of evolutionary biology is, therefore, to explain how high-fidelity replication originally evolved, and what selective pressures now maintain it. As part of a solution to this problem, I present a simple “Everest hypothesis”. This proposes that natural selection consistently adds unnecessary complexity to the mechanisms that transfer genetic material between individuals. Many sexual organisms choose their mates (I suggest) by monitoring a variety of complex of behaviours, physical displays and biochemical mechanisms, often generated by the interaction of many gene products acting together or in sequence so that a defect in a single gene can result in failure to accomplish genetic transfer. Individuals with defective DNA polymerases are likely to have more mutations in these (and all other) genes, and, since most mutations are deleterious, the chance of transferring genes that encode error-prone polymerases is reduced. Many puzzling biological phenomena among sexual organisms can be explained along these lines. The migration and spawning of Atlantic salmon and the complex displays of birds of paradise may, for example, be best understood as “tests” to establish whether potential sexual partners are capable of high-fidelity genetic replication. (Other explanations of these phenomena in the scientific literature may be correct but less important.) Animals that have developed physical handicaps that appear to be harmful, such as peacocks, and animals that undertake remarkable migrations, such as arctic terns, may be special (extreme) cases. I also present suggestions for experiments to test the hypothesis.
Introduction
Imagine a woman who announces publicly that she will have sex with any man, but only on the summit of Mount Everest. Moreover, the potential partners must have solved a difficult sudoku puzzle that they pick up on the way up and (so that she can choose quickly) they must write – display – their solutions in large numerals on a banner that they must bring along. If it were practical this would be a reasonable mating strategy for both partners: both mother and father are likely to have better-than-average genes. I suggest in this essay that many plants and animals use similar strategies: they set up challenging practical “obstacle courses” for potential mates, and may also demand complicated physical displays that can only be generated by the interaction of many genes. These strategies can signal to individuals that potential partners have genes that are of good quality, and, in particular, that are capable of high-fidelity genetic replication, before they agree to mate with them.
The problem
No molecular replication system is error-free. The mutation rate in humans has been estimated to be around 2.5 x 10-8 mutations per nucleotide site per generation [Nachman] and bacteria have similar mutation rates (approximately one error per billion base pairs copied [Peck]). DNA replication (and RNA replication in RNA viruses) is carried out by proteins, which are themselves encoded by DNA or RNA sequences. There is therefore a chance that mutations will occur in the genes encoding DNA polymerases and their many “helper” proteins. While some of these mutations will have no effect, and some will reduce fidelity to an extent that is fatal, some must inevitably reduce fidelity to a small degree. Note that such mutator mutations must exist, and, of course, they increase the chance of other mutator mutations appearing during development or in future generations. This might result in a slow – or rapid – increase in mutation rates, eventually causing an “error catastrophe” that would kill the individual. Biological strategies are therefore needed to avoid this fate, so that simple life-forms can persist and complex life-forms can evolve from them. This essay aims to identify behavioural, physiological and biochemical strategies that can reduce the number of low-fidelity lineages in a population.
Sexual reproduction
Sexual reproduction is extraordinarily ubiquitous – different forms of it are used by virtually all plants and animals, most fungi, and many protists. Some biologists regard sexual reproduction as paradoxical [Otto]. Many theories have been put forward to explain the ubiquity and persistence of sex. Hill and Robertson suggested that sex allows the combination of two or more beneficial mutations in one individual, allowing more effective selection [Hill]. Similarly, two or more deleterious mutations can be combined, accelerating their removal from the population [references in this section are mainly from https://en.wikipedia.org/wiki/Evolution_of_sexual_reproduction ]. Ronald Fisher suggested that sex might allow advantageous genes to escape their genetic surroundings if they happen to arise on a chromosome with deleterious genes. Heng suggested that sex can weed out major genetic changes such as chromosomal rearrangements, but permit minor variations such as nucleotide alterations. A widely-discussed theory, known as the Red Queen Hypothesis, suggests that sexual lineages are better able to resist parasites, because the combination of parasitic resistance alleles of offspring will differ from their parents’ [William D. Hamilton]. Others see sexual reproduction as a DNA repair mechanism [no ref in Wikipedia]. On the other hand, sex has disadvantages. John Maynard Smith pointed out, for example, that an asexual population can grow much faster than a population with two sexes because males do not produce eggs and bear offspring. Serious problems with the conventional explanations of sex have led many biologists to conclude that the benefit of sex is a major unsolved problem in evolutionary biology [so says Wikipedia; reference? Maybe William D. Hamilton again].
I suggest that many of the theories of sex discussed above are important and correct, but that the most fundamental and important aspect has been overlooked. So, to all these explanations I would like to add one more: that sexual reproduction is a very effective way to recombine lineages that have acquired beneficial mutations while preserving something that is very valuable but easily lost – high-fidelity replication.
Mate selection in sexual reproduction allows low-fidelity lineages to be avoided
The only phenotype of mutator mutations is (presumably, see below) an increased number of mutations, which appear randomly throughout the genome. How can this phenotype be detected? I suggest that organisms use a variety of approaches, which monitor complex behaviours or structures that are generated by many genes. For example, many finely-tuned gene products must be required to make the feathers of a cock bird of paradise, while other genes generate the complex behaviour to display them effectively. Still other genes allow a female bird of paradise to identify the “correct” feathers and display. Mutations in any of these genes could prevent mating. Some animals have complex features that attract mates but are positive encumbrances, such as the tail feathers of a peacock. Biologists put these features, which seem to be harmful for the species as a whole, down to sexual selection, meaning that they are the result of a self-reinforcing “fashion” among the females – it is said that any particular female can’t easily break away from this harmful fashion because if she produces male offspring without these features they will not be able to find mates. Note, however, that not all birds have exotic feathers – many are plain, with both sexes looking alike (for example blackbirds). However, birds that lack complex plumage often have complex vocalisations, which may serve a similar purpose. Humans are attracted to partners with athleticism, pretty faces (which are close to, but not identical to, average faces [Nature paper]), and intelligence and/or a sense of humour, both of which are the product of an extraordinarily complex organ – the human brain. Other animals go to extraordinary lengths to migrate in order to breed. For example, birds often undertake dangerous migrations to breed in remote locations. “Breakaway” populations that either do not migrate or migrate less far often exist, but they do not generally outcompete the populations that complete the longer migrations. Atlantic salmon are able to migrate from fresh water to the ocean, and then return, with both sexes undertaking dangerous journeys, including adapting to changing salinity, leaping up waterfalls, avoiding predators, and swimming in shallow water, to return to the streams where they were bred in order to mate. (This really is similar to the fictitious woman who would be willing to mate on the summit of a mountain.) Presumably lineages have appeared in the past that bred in less demanding freshwater or saltwater locations, but, I suggest, they didn’t thrive because they lacked this very effective strategy for eliminating individuals with more mutator mutations and slightly higher mutation rates. A similar argument can be applied to the extraordinary migrations of birds. Invertebrates may also use complex features and behaviours to attract mates. For example, fireflies receive and transmit flashed encoded messages to attract mates, while medflies and some spiders perform complex dances. Insects such as cicadas, mayfly and ants lack wings throughout most of their lives, but grow wings – very complex structures – in order to mate. (I appreciate that wings also allow such insects to disperse themselves, but I suggest that they serve a double purpose – allowing dispersal while also acting as a filter that removes low-fidelity lineages. Note that selective pressures during the wingless phase may encourage the loss of wing genes.) The North American migrations of monarch butterflies are another, extraordinary, case. Each year, populations east of the Rocky Mountains complete a dangerous multi-generational migration between overwintering sites (the largest being as far south as Michoacán in Mexico) and their northern breeding grounds, mainly near the Great Lakes. Since monarch populations in other regions do not migrate, it is at first sight unclear why North American populations complete such dangerous and complex migrations, requiring four generations to complete the cycle. The Everest hypothesis can provide an explanation, which is that individuals that participate in the migration are more likely to find mates that are capable of high-fidelity replication. (Since the cycle is multi-generational, many of the genetically-encoded behaviours and physiological changes involved cannot be conserved by selection en route in low-fidelity lineages.) Corrals may provide another example, since they monitor water temperatures, light, and the cycles of the moon (or tides) in order to synchronize their spawning.
Zahavi’s handicap principle
In 1975 Amotz Zahavi suggested that characteristics, behaviours and structures in animals that confer handicaps may evolve by sexual selection because they “test the quality” of the animals that possess them [Zahavi]. The characters selected in this way must, according to Zahavi, “lower the fitness of the selected sex in relation to the main ecological problems of the species” [Zahavi]. Like the Everest hypothesis, the handicap principle suggests that sought-after characters are used to advertise the quality of genes whose effects would otherwise be hidden. There are, however, fundamental differences between these two hypotheses, listed in table 1. As Zahavi pointed out, the evolution of these sought-after characters may be explained by more than one hypothesis [Zahavi]. Moreover, I’m certainly not saying that the handicap principle is wrong: in the example given in the Introduction, above, if a man arrived on the summit of Mount Everest with a bunch of flowers, he would be applying the handicap principle. It might work! I suggest, however, that the Everest hypothesis provides a simpler and more universal explanation of the many surprising mechanisms that are involved in the reproduction of plants and animals.
|
Handicap principle |
Everest principle |
| The principle proposes evaluation by individuals of an indirect signal: mates that squander scarce resources show that they are of “good quality” and are selected. | The principle proposes direct evaluation of characters that are correlated with low mutation: individuals simply select mates that have a sought-after character that can only be created by an intact set of genes. |
| This eliminates bluffing by giving a reliable signal that cannot be faked because it requires the consumption of a scarce resource. | Bluffing is impossible because the sought-after character can only be generated by the possession of the appropriate genes. |
| Seeks to explain the puzzling appearance and behaviour of some animal species. | Seeks to explain the ubiquity of sexual reproduction and the persistence of high-fidelity genetic replication. |
| Applies to a subset of animals that have developed handicaps. Major handicaps seem to appear randomly in a subset of species. | Applies to all complex organisms. In some cases, the sought-after characters may be handicaps, but in other cases they may be useful in themselves (e.g. intelligent brains). |
| The signal evaluated should lower the fitness of selected individuals “in relation to the main ecological problems” of the species. | The signal evaluated may be costly to the individual. However, it may also be beneficial, such as the possession of an intelligent brain, or strong muscles. |
| Patterns such as the eyes on a peacock’s tail are incidental – the cost of the sought-after character is the important factor. | Sought-after characters include (but are not limited to) patterns that can only be generated by the interaction of many genes. The extraordinary symmetry of the peacock’s tail suggests that symmetry is strongly selected by peahens (Figure 2.1). |
| The principle focuses on biological fitness, which is difficult for scientists to define or quantify. For example, if the environment changes a different set of alleles may instantly confer greater fitness. | The principle focuses on mutation rates, which are well-defined and can be measured directly by scientists [Peck]. |
| Predictions: | |
| Species and populations that have greater handicaps are expected to be at a disadvantage compared to other comparable groups with more modest handicaps. | Species and populations with more extreme handicaps may be at a long-term selective advantage compared to other comparable groups, and may thrive, if the handicaps successfully reduce the transmission of error-prone polymerase genes. |

Figure 2.1. The extraordinary symmetry of a peacock’s tail. In 1975, Amotz Zahavi proposed the “handicap principle” to explain the evolution of elaborate features such as these tails. The principle suggests that by squandering scarce resources by growing and maintaining such features, peacocks and other animals show potential mates that they are of “good quality” and are selected. However, the handicap would be almost identical without the elaborate markings, suggesting that they have some other benefit. The Everest hypothesis notes that a well-formed tail shows that the peacock has an intact set of genes for constructing this feature, suggesting a low mutation rate. Similarly, a female that recognises a well-formed tail has an intact set of genes for tail-recognition.
Flowering plants
At first sight it might appear that flowering plants are a problem for the theory. However, it turns out that plant fertilization is complex: pollen tube elongation in the maternal tissue and navigation to the ovule require intimate successive cell–cell interactions between the tube and female tissues. This process can create complex “tests” for pollen grains (which should be thought of as haploid organisms that are capable of producing sperm) using multi-layered signalling pathways that involve many gene products, which may weed out error-prone lineages (Figure 2.2, from Li et al.). Thus the Everest hypothesis can explain why complex multi-layered mechanisms are beneficial.
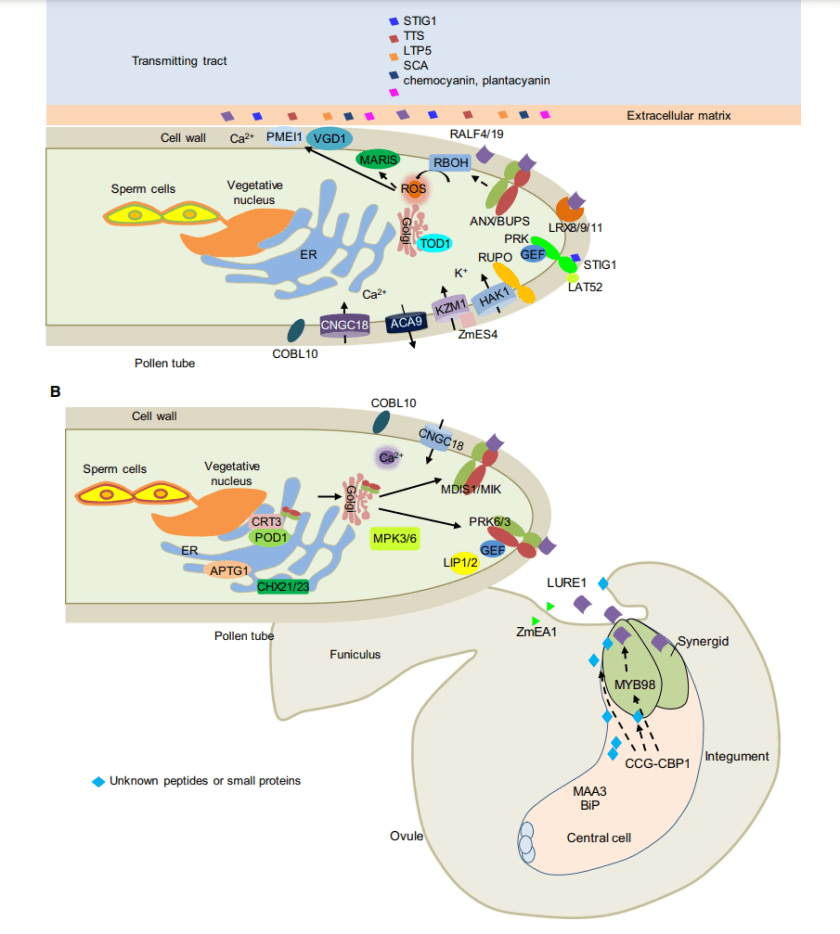
Figure 2.2. Fertilization of flowering plants is highly complex, involving multilayered signalling pathways, with many gene products that are expressed in both pollen and the female tissues. The Everest hypothesis suggests that this complexity reduces the chance that low-fidelity strains will successfully reproduce.
Other benefits of complex mate selection
Note that the mate-selection strategies discussed above can also filter out other defects that would otherwise be hidden. Complex behaviours, displays and biochemical mechanisms can also show up mutations in “house-keeping” genes that are active in all cell-types such as ribosomal and cell-cycle proteins, histones, mitochondrial proteins, as well as transcription, protein processing, RNA splicing, and translation factors.
Plant and animal breeding
It would be very interesting to talk to breeders. Do they notice that certain strains (although they may have desirable characteristics) are “weak”? Can the weakness be eliminated by crossing with more vigorous strains? I know that foresters take a lot of trouble to acquire good-quality seed. Rose breeders sometimes refer to “effete” lineages, which have difficulty reproducing. Isolated animal communities in zoos and laboratories could be studies, including sequencing individuals’ polymerases. Can isolated communities maintain their fidelity by preventing low-fidelity individuals from breeding?
Early-expressed suicide genes etc.
I can imagine a complex biochemical mechanism that is highly sensitive to mutation, operating in early development. Mutation would result in the death of the organism. This would be the biochemical equivalent of asking a baby to walk across a high tight-rope (if that were possible). Do such things exist? Just an idea – something to look for.
Chromosomal abnormalities are found in more than half of embryos miscarried in the first 13 weeks [Kaji 1980] – this may be related to my suggestion. And why did the other half miscarry?
Suggestions for experimental and observational testing of the Everest hypothesis
Several scientific approaches could be used to test the Everest Hypothesis. Similar studies could be performed with any convenient sexual organisms, including yeasts, protists, insects, flowering plants, fish, mice, birds or mammals (possibly in captivity, for example in zoos.) I suggest experiments along the following lines: (1) sequence PAPs from wild organisms in large populations, which can be assumed to be high-fidelity. (2) Identify or create inbred populations and sequence their PAPs. Identify inbred lineages with mutant PAPs, which will often show increased mutation rates. (3) Set up new colonies, starting each colony with single pairs of organisms. Sequence PAPs and a selection of other genes to identify high-fidelity and low-fidelity colonies. (4) Quantify and compare the health of high and low-fidelity colonies. This can shed light on the expected prevalence and evolution of low-fidelity lineages in nature. (5) Now introduce high-fidelity individuals to low-fidelity colonies, and low-fidelity individuals to high-fidelity colonies; use sequencing to compare the rates at which the two classes of PAP genes invade their respective colonies. The Everest hypothesis predicts that high-fidelity PAP genes will replicate and spread faster. A second experimental approach would test whether the application of strong selective pressures encourages the emergence of low-fidelity lineages. For example, colonies could be sustained on unsuitable foods, or exposed to toxic compounds. Novel behaviours could also be selected, for example by eliminating drosophila and other insects that are attracted to electrical insect killers with UV lamps. Evolutionary theory suggests that low-fidelity lineages will be more prevalent after strong selection and rapid adaptation. A third approach is observational. Since the Everest Hypothesis suggests that long migrations are an effective way to eliminate mutator mutations, it predicts that migratory lineages will infiltrate non-migratory populations more often than the reverse. This prediction could be investigated in monarch butterflies by constructing phylogenetic trees based on monarch sequences. Another approach would compare the importance of symmetry to that of other features. For example, if the tail of a peacock that was known to be highly attractive to peahens were to be painted so as to reduce the symmetry, to what extent would it reduce the attractiveness of its owner (has this experiment already been done?).
Part 3: the Preservation of Replicative Fidelity in Quasispecies (First Draft)
Abstract
Mate selection is not available to simple biological entities such as viruses, but recombination between strains can still eliminate many undesirable mutator mutations. I suggest that the fidelity of replication in viruses varies by orders of magnitude depending on selective conditions. Strong selective pressures may favor lower-fidelity strains. The periodic surges of cases of viral diseases such as influenza and Covid-19 may reflect the emergence of low-fidelity strains, which can adapt to new conditions quickly, but then collapse due to the accumulation of mutations in essential viral genes, including polymerase genes. New variants (such as Alpha, Delta, Omicron etc in SARS-CoV-2) may emerge as a result of recombination between low-fidelity strains with desirable mutations (including mutations in surface protein genes) and high-fidelity strains that are capable of accurate replication. Zoonoses and the origin of Covid are considered. Finally, a thought-experiment suggests that individual polymerase genes with greater fidelity may evolve during extended periods of low evolutionary change i.e. in populations whose genes are close to equilibrium.
Mutator mutations in viruses
Biological entities such as bacteria, archaea and viruses are relatively simple, and they are often asexual, existing as quasispecies (large groups or “clouds” of related genotypes). They therefore have limited or non-existent opportunities for mate selection. This essay will focus on the simplest group, viruses, including SARS-CoV-2. Although asexual, viruses can recombine when two viruses infect a cell simultaneously, with the result that part of the genome of progeny virions comes from one lineage, the rest from another. New variants are therefore often the result of one or more recombination events [Bill Gallaher].
Coronaviruses have their own polymerases, which replicate their genetic material, RNA. They also have some of the largest RNA virus genomes (around 30,000 base pairs) and they are said to exist close to “error catastrophe”, where a small increase in the mutation rate would destroy the virus. They need high-fidelity polymerases to maintain such a large genome. For example, SARS-CoV-2 uses five non-structural proteins to construct the complex that replicates its RNA (Figure 3.1). (When I refer to “polymerase” in this essay I have in mind the whole complex, including all the proteins that make it up.) This complex includes a proof-reading function (comprising NSP14 and NSP10) that reduces mutation by a factor of around 20. All these proteins can mutate, which must occasionally give rise to low-fidelity lineages. The existence of low-fidelity lineages is thus not a matter of conjecture – they must exist. It should be a high priority to find out how common low-fidelity SARS-CoV-2 lineages are, and to determine their role in viral evolution.
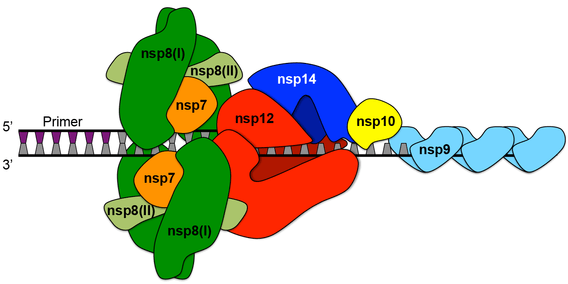
Figure 3.1. The multi-protein replicase-transcriptase complex of coronaviruses. Mutations must inevitably occur occasionally in all these proteins, and most mutations are expected to be deleterious, implying that they will usually reduce replicative fidelity.
Quasispecies swarms
I suggest that in nature the fidelity of viral quasispecies swarms varies over several orders of magnitude. (1) The most stable lineages are those that persist over years with low mutation rates. I suggest that high-fidelity strains are required to establish diseases that can persist in new host species. High-fidelity strains are depicted by brown lines on Figure 3.3. (2) Strains with intermediate fidelity (blue lines in Figure 3.3) persist long enough to cause limited epidemics, possibly infecting millions of people, but they eventually become extinct. Their survival is limited by the mutations that inevitably accumulate in their essential genes, including their polymerases and associated proteins. (3) The lowest fidelity lineages probably arise and persist only in individual hosts. Since they are unstable, scientists might not notice them unless they look for them. They evolve rapidly and so are likely to lose the functionality of genes that are essential for transmission between hosts. They may, however, (I suggest) be highly pathogenic, possibly being responsible for many deaths caused by viral illnesses. It is likely that type 1 viruses that are passaged through cells that they are not adapted to often lose fidelity, becoming type 2 and 3 strains. Type 3 strains may be difficult to propagate for more than one or two passages.
Unexplained features of Covid-19 epidemics
The extraordinary surges and rapid collapses of cases that we have sometimes seen in Covid-19 are some of the pandemic’s most puzzling features. For example, dramatic surges and collapses were seen in both South Africa and India (Figure 3.2A), two countries where lockdowns may be relatively ineffective. The curves seen are quite unlike the curves generated by Gompertz functions, which are the expected result of increasing immunity. A Gompertz curve has a flattened, rounded summit, whereas Covid-19 cases often show rapid monotonic rises followed by sharp reversals and monotonic falls. Similar patterns can be seen in many countries, including the UK, France, Indonesia and Austria (Figure 3.2 B-D). I suggest that these surges mainly comprise low-fidelity strains, because their higher mutation rates allow them to evolve faster than the ancestral high-fidelity lineages. According to this view a high mutation rate in the presence of strong selection is favourable at first, then detrimental. It should be borne in mind that mutations that increase the chance of transmission will often not be selected within the host – in fact the opposite may be the case – so many advantageous mutations can only be selected during transmission. Moreover, most infections are caused by the transfer of a relatively small number of virions that are airborne. So, with more mutations, and strong selection, it seems likely that more error-prone mutants can adapt to new opportunities more quickly than very high-fidelity lineages. Such lineages, however, have (I suggest) no long-term future because they are prone to error-catastrophe (unless they can recombine with high-fidelity lineages, see below). The overall effect would be alternating surges and collapses of cases, as shown in Figure 3.3.
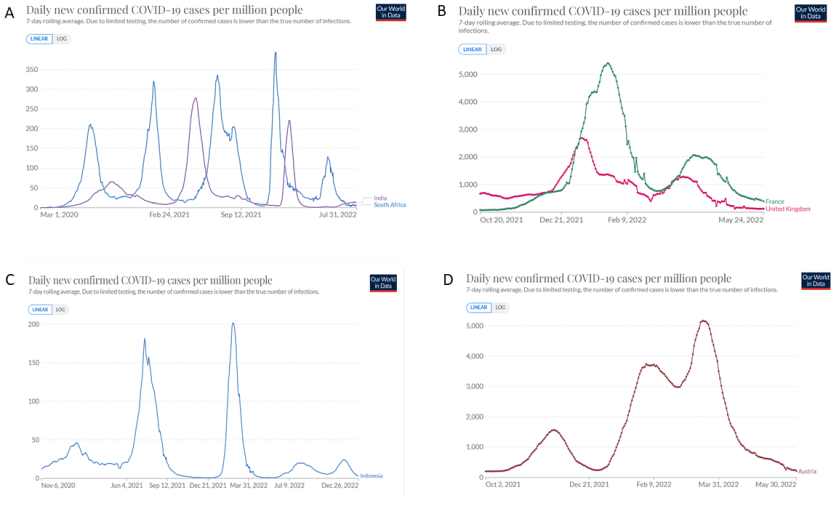
Figure 3.2. Covid-19 surges that were followed by sudden collapses of cases in several countries. A: Indonesia, B: France and the United Kingdom, C: Indonesia, D: Austria.
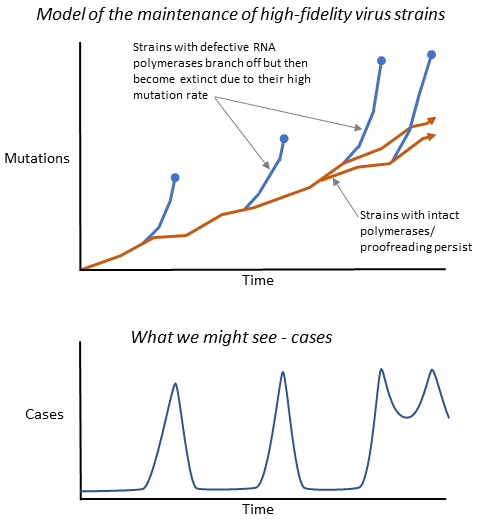
Figure 3.3. A schematic model showing how strains with defective, low-fidelity replicative machinery (blue lines) might evolve faster, giving them a short-term advantage. However, the model suggests that mutations will accumulate in essential genes, so such strains will be unable to compete and survive longer-term, and will normally become extinct. High-fidelity strains (brown lines) can persist, however.
According to this analysis, we expect to see an increased number of mutations in the data produced by organizations such as http://Nextstrain.org during cases surges, with a reduction at the end of the surge when high-fidelity strains reappear. This is not seen (although, as expected, new variants tend to start with more mutations than their predecessors, indicated by the red arrow on Figure 3.4). Remarkably, there is a plausible explanation for the observed lack of excess mutations during surges: NextStrain has a policy of excluding any strain with an unusual number of mutations that differ between the query sequence and the nearest neighbour sequence (they refer to these as “private” mutations.) It is often noticed, however, that when new variants appear more mutations are seen [R. Neher, communication on the Nextstrain discussion forum]. This is generally put down to problems with the amplification schemes used, but it could also reflect a real increase in the number of mutations that arise during surges. In other words, scientists may have seen the effect that I am postulating, but misinterpreted it.
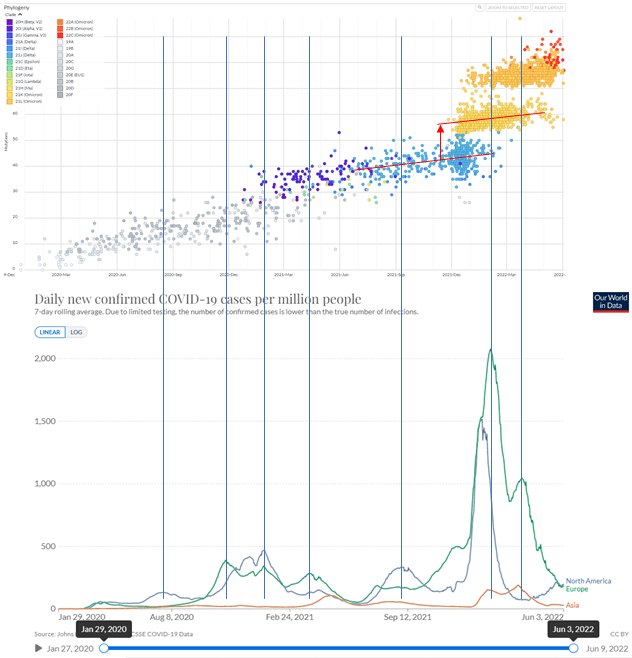
Figure 3.4. Covid-19 sequence databases such as NextStrain do not show increased mutation during surges of cases, although new variants tend to have more mutations (red arrow). This can, however, be explained by the policy of NextStrain to exclude any strains from the database that have more than the normal number of “private mutations”, which are mutations that are not shared with any other strain. See the main text for more details.
Several other explanations of the strange peaks and falls in cases have been proposed, such as increasing immunity, behavioural changes in hosts, and non-linear percolation effects. Taking these in turn, (1) increasing immunity cannot plausibly explain, for example, the collapse of cases at the end of e.g. the first surge in South Africa: it is clear that immunity in the population at the end of the surge was not high because it was followed by several larger surges (Figure 3.5). Moreover, we would expect the curves to flatten off gradually and the tails to be longer if the shape was driven by the slow increase of immunity. (2) Behavioural changes are not well-correlated with cases. For example, in December 2021 cases in South Africa first surged, then collapsed, although mobility increased rapidly for a week after the peak (Figure 3.7). A similar pattern was seen in Europe (Figure 3.8: cases and mobility in Austria are shown because the surge was not close to the Christmas holiday; cases close to holidays are difficult to interpret). (3) It has been suggested that non-linear percolation effects can explain the peaks seen, but it’s hard to see how they could routinely generate monotonic rises, followed by monotonic falls. Figure 3.9 shows the patterns that would be expected in an extreme case of non-linearity. Here (red curve), data from the London stock exchange has been increased or decreased by 500 points when it crosses two thresholds.
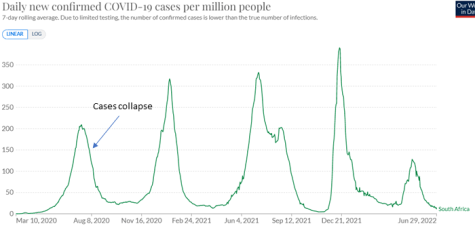
Figure 3.5. In late July 2020, Covid-19 cases in South Africa collapsed abruptly. This could not be explained by high levels of immunity in the population, because this surge of cases was followed by four similar surges during the next two years.
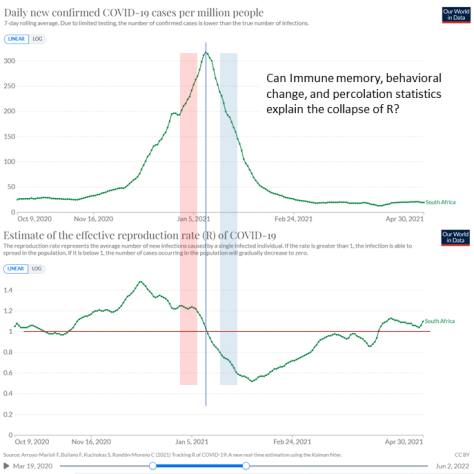
Figure 3.6. In less than a month, the effective reproduction rate (R) of Covid-19 in South Africa collapsed from around 1.2 (pink band) to below 0.7 (blue band). Conventional explanations struggle to explain this sudden change.
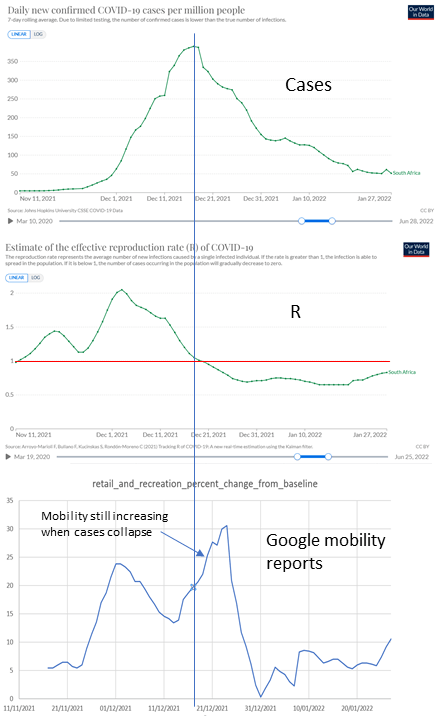
Figure 3.7. After Covid-19 cases in South Africa suddenly began to fall in December 2021, mobility measured by Google reports continued to rise strongly for a week.
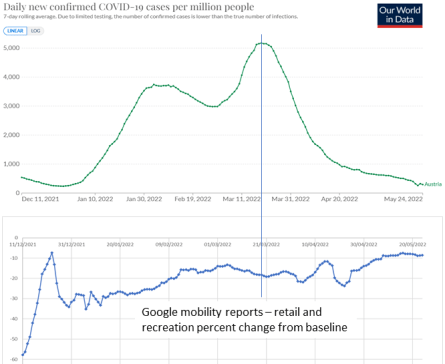
Figure 3.8. Covid-19 cases collapsed in Austria in March 2022. During this period, mobility was almost unchanged as shown by Google mobility reports.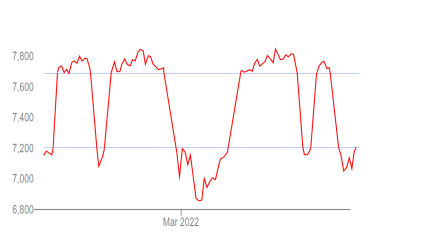
Figure 3.9. It has been suggested that non-linear percolation effects could explain the rapid surges and collapses of cases that are observed. However it is hard to see how they could generate so many, very sharp, peaks. This figure shows typical curves might be generated by extreme non-linearities. Here I have increased and decreased data from the London Stock Exchange (red curve) by 500 points whenever the curve crosses upper and lower thresholds (blue lines).
The Omicron variant and other SARS-CoV-2 variants of concern
Omicron (BA.1) has puzzled virologists because it has 29 non-synonymous mutations in the spike gene, but only 15 non-synonymous mutations in the whole of the rest of the genome. Moreover, an anomalously low proportion of the mutations in the spike of Omicron (and other variants) were caused by C-to-T nucleotide transitions. Most C-to-T transitions in SARS-CoV-2 are thought to be generated by host modifications to viral RNA [], and we can get an idea of the underlying frequency of C-to-T mutations by looking at synonymous mutations, which are not expected to be strongly selected. Combining the totals of all synonymous “defining mutations” of Alpha, Beta, Gamma, Delta and Omicron variants, the majority (53%) were C-to-T transitions, as shown on table 2 [data from NextStrain.org]. However, only 11 out of 92 non-synonymous spike mutations (i.e. 12%) were C-to-T transitions [data for Beta, Delta, Lambda and Omicron came from a twitter-acquaintance. Can anyone help me to get more consisten data – e.g. filling in the question marks in the table?]. This data is summarized in Table 2.
| Length | Synonymous mutations~ | Of which, C-to-T~ | Synonymous C-to-T mutations, %~ | Mutations* | Of which, C-to-T* | C-to-T mutations, %* | |
| ORF 1ab | 21,287 | 21 | 14 | 67% | ? | ? | ? |
| “Structural” proteins | 8,138 | 21 | 8 | 38% | ? | ? | ? |
| Spike | 3,849 | 1 | 1 | 100% | 92 | 11 | 12% |
| ~From “defining mutations” of Alpha, Beta, Gamma, Delta, Omicron, from Nextstrain.org | |||||||
| *From sequence data from Beta, Delta, Lambda, Omicron | |||||||
I suggest that these variants were generated by recombination between error-prone strains (which had acquired many beneficial, random, non-C-to-T mutations via defective polymerases) and more stable high-fidelity strains. This mechanism is illustrated schematically in Figure 3.10. I suggest that much of the right-hand-end “structural protein” sections of the genomes of these variants, including the spike genes, came from error-prone partners, while much of the non-structural protein sections, including the polymerases, came from high-fidelity partners.
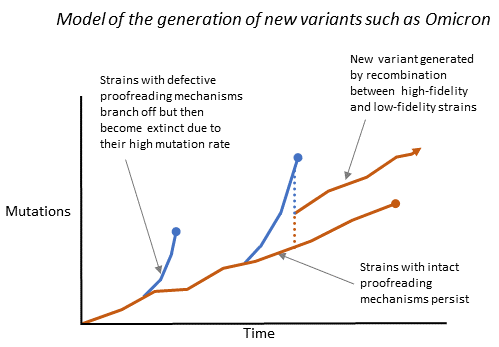
Figure 3.10. A schematic model showing how the recombination of low-fidelity strains with beneficial mutations (blue lines) with high-fidelity strains (brown lines) might generate stable strains with many beneficial mutations.
The Speculative nature of the rest of this essay
What follows is probably even more speculative than the preceding sections. Experiments would be very helpful. Or does anyone know of any existing data can shed light on the mutation rates of viruses after they are introduced to new hosts or cell-lines?
Sniffles, coughs, sinusitis, bronchitis and bronchiolitis
The expected patterns of selection of quasispecies suggests the following analysis: successful infections of the nose and throat by respiratory viruses are expected to be caused by relatively high-fidelity lineages. However colds sometimes develop into sinusitis, bronchitis or bronchiolitis – sometimes after the cold seems to be improving. The downturn may reflect the appearance of new forms with reduced fidelity, which in turn leads to reduced thermal sensitivity, allowing the infection to move inwards or downwards (Shaw Stewart and Bach, 2021). This analysis can explain why respiratory infections are often passed on in the early stages of an illness, and why patients may be less infectious in the later stages of their illness, despite suffering from severe sickness.
Variolation
Variolation was the earliest form of immunisation against smallpox. Material from smallpox scabs or fluid from pustules was rubbed into scratches on the skin of the individual being immunised. It has been suggested that this led to a milder form of the disease because it was localised to the site of inoculation. Another explanation is that the virus that is present in scabs and pustules at the end of the illness is expected to include many low-fidelity lineages that are less dangerous than the lineages that would typically be transmitted at the start of the illness.
Selection in new hosts
Consider the case of a virus spilling over to a new host and starting to replicate. A strong selective pressure is immediately applied. There are now many sites for potential mutations that could allow the virus to bind more strongly to host receptors, enter cells more efficiently, interact better with host proteins, replicate faster, avoid the new set of immune defences etc. It would, however, take a long time to sample these mutations with a high-fidelity polymerase complex. The most efficient way to accumulate many beneficial mutations in a single lineage is to mutate the viral polymerase first, increasing the error rate, then allow the virus to adapt to its new host. However, high fidelity cannot re-emerge – that would take eons of slow selection. So this lineage has no future on its own – mutations will steadily accumulate in vital functions and it will go extinct.
If, however, a high-fidelity virus co-infects the host (and it could be a new sample of the original strain) then recombination can give the best of both worlds – a well-adapted virus with a low mutation rate.
The origin of stable laboratory influenza strains
The existence of stable laboratory influenza strains might at first appear to be a problem for this analysis, since viruses ought (according to the analysis) to develop high mutation rates when they adapt to new hosts such as eggs or cell cultures. This might be expected to prevent the strains from replicating indefinitely in their new hosts – they might be expected to accumulate fatal mutations. However, laboratory strains such as PR8 (isolated in Puerto Rico in 1934) and WSN (isolated in the United Kingdom in 1933) appear to be completely stable in the lab. There are several important points to be made here: (1) since the introduction of reverse genetics systems in the early twenty-first century, the stability of virus cultures is no longer a problem, because stable sequences can be synthesised, cloned and re-used indefinitely. (2) Only a few stable laboratory influenza strains exist [please correct me here if I’m wrong!]. (3) It is often difficult to establish stable cultures of new viral isolates in the lab. Passaging experiments often seem to be successful at first, but cultures become negative after several transfers [e.g. Francis]. (4) Stable strains such as PR8 and WSN may be the result of accidental recombination. Note that contamination is a constant problem when working with viruses. For example, standard laboratory protocols recommend sterilizing the shells of eggs before using them. PR8 and WSN were maintained in embryonated eggs for many years and are presumably well-adapted to that host. If the progenitor strains became adapted to eggs (but lost fidelity in the process) they might have regained fidelity by recombination with a freshly-isolated strain. The result might be a strain that was stable in eggs in the long term.
Note added 15/01/2023: thinking about it, unless steps are taken to prevent cross-contamination it is likely that defective laboratory strains will often be “rescued” by spontaneous recombination (or reasssortment in influenza) with intact strains. Consider, for example, the controversial experiments by Ron Fouchier where the dangerous H5N1 avian influenza virus was repeatedly passaged in ferrets, resulting in airborne transmission in ferrets [Herfst]. If a few lineages acquired mutations in ferrets in their polymerases that resulted in low-fidelity replication, lineages might soon appear with several advantageous mutations in the host receptor-binding protein hemagglutinin. These might out-compete the original strains. If, however, strains with intact polymerases were to be accidentally transferred to the ferrets (e.g. via aerosol or fomite transmission) reassortment might take place without the experimenters even being aware of it, such that the three segments encoding (intact) polymerases were picked up. The result might be a high-fidelity strain with adaptive mutations in genes such as the hemagglutinin gene.
Zoonosis and the origin of Covid-19
Every year, there are roughly 500,000 cases of Lassa fever in West Africa, including around 5,000 deaths. People usually acquire this illness through contact with the urine or feces of the Natal multimammate mouse. Other viruses, including the Nipah, Lujo, Ebola, and Marburg viruses, regularly spill over to humans, mostly from bats or rodents, sometimes causing hemorrhagic fevers with high case fatality rates. However, in spite of hundreds of thousands of apparent opportunities for pandemics to begin each year, they are rare. I suggest this is because a virus needs a high-fidelity polymerase to become permanently established in a new species (see above). Strains with inadequate fidelity may persist for a few weeks or months before they inevitably become extinct. (SARS-1 may be an example of this.) Since the strong selective pressures that a virus encounters when it replicates in a new host are likely to favour low-fidelity polymerases that can provide plenty of mutations, most viral spill-overs to humans and other animals are (I suggest) self-limiting.
Several steps are therefore required (I suggest) to establish a permanent new human viral disease. A virus must first spill over to humans, then begin human-to-human transmission so that it can adapt to its new host (usually losing fidelity in the process). Next, the virus needs to co-infect a single host (actually, a single cell) with a related virus that has retained high fidelity, in order to recombine with it. There are two possibilities here: either the new arrival could recombine with an existing human virus by co-infecting with it; or the original animal virus might spill over again from its previous host, co-infecting a human with the low-fidelity, human-adapted strain. Both possibilities might result in a well-adapted, stable strain. I suggest that the rarity of such co-infection plus recombination is what protects us from pandemics in spite of hundreds of thousands of zoonotic infections occurring every year.
Figure 3.11. How repeated spillovers may give rise to stable strains through recombination.
Incidentally, this analysis can explain something else: why animal viruses rarely spread from person-to-person. This is a surprising observation – you might expect that a viral lineage has already been selected to infect humans would be more likely to infect other humans than the original strain. I suggest that low-fidelity lineages rapidly emerge during the first infection, and they are unable to infect other humans because they have lost vital functionality, including functionality that may be involved in transmission.
Since it has persisted in humans for three years at least, Covid-19 clearly includes high-fidelity strains. There are many ways in which high-fidelity strains could have arisen. I am listing a few here, not in any particular order.
Scenario 1: SARS-CoV-2 may be a product of repeated spill-overs from bats to humans, probably in Southern China, which eventually gave rise to a human-adapted, high-fidelity strain by recombination. I suggest that a bat virus would first have to infect humans and become adapted to them, then recombine with a similar bat strain after an independent spill-over to create a high-fidelity strain. Humans with antibodies to SARS-related viruses were found in Yunnan before Covid-19 began [Wang], and we know that EcoHealth Alliance was actively looking for novel human coronaviruses in Southern China [This Week in Virology podcast (TWiV) 615, minute 28]. A possible route for SARS-CoV-2 to reach Wuhan is that it was taken there by scientists. But why did the pandemic first become apparent in Wuhan rather than e.g. Southern China? One possible answer is that scientists engineered features such as the furin cleavage site in the spike protein, which increased the transmissibility of the Wuhan strain.
Scenario 2: another possibility is that a bat virus was brought to a lab and repeatedly passaged through human cells or humanized mice. This might have increased its adaptation to humans. However, it is (in my opinion) unlikely that it could have become fully-adapted to humans in cells or mice because in addition to gaining entry to cells and replicating it needs to avoid human immune responses and be transmitted efficiently by the respiratory route. The virus may therefore have also infected laboratory workers or other people connected to the laboratory. After this it would have to coinfect an individual and recombine with a high-fidelity strain, which could have been any related freshly-isolated bat virus in the lab.
Scenario 3: as in the first scenario, above, a novel human-adapted high-fidelity virus might have arisen from repeated spill-overs to humans in for example, Southern China. This virus began to spread in Southern China but was not noticed. It somehow reached Wuhan, where it naturally mutated and became more transmissible.
Scenario 4: a virus spilled over enough times from animals to humans in Wuhan to allow recombination, for example at a seafood market. This seems less likely because the exposure of humans to bat viruses in Southern China is much greater, yet this exposure very rarely causes human epidemics. The scenario where the virus spreads from bats to a third species, then from that species to humans, is problematic because of the need to re-establish high fidelity by at least one recombination event, or, more likely, two. (SARS-CoV-1 may not have the capacity for high-fidelity replication, which could have limited its persistence in humans.)
Suggestions for experiments to test these ideas
I can think of dozens of experimental approaches to test these ideas. I’m going to add suggestions here as the occur to me.
- Use nanopore sequencing to see how fast viruses mutate when they infect new hosts such as cell cultures in the lab. My hypothesis suggests that after adaptation to cells their mutation rate will be higher than the wild-type virus’s.
- Use nanopore sequencing to compare sequences taken from the airways at the start of a normal infection to those taken from the internal organs of a very sick patient. My hypothesis suggests that the latter will have a much higher mutation rate.
- Sequence the polymerase of a wild virus, then passage the virus through cells several times. Sequence the polymerase after passaging and construct reverse genetics systems with both polymerases. Compare their fidelity.
Perspective
An example in the form of a thought-experiment may shed light.
Imagine a laboratory with three fermenters that are continuously fed with a suspension of monkey cells that never varies, and from which waste products are continuously removed. The scientist running the lab adds a human RNA virus to the fermenters. This virus has (say) ten genes, including its own RNA polymerase.
The scientist monitors the fermenters every day, and sequences strains as appropriate. During the first week the number of virions increases steadily in all fermenters, as the virus adapts to the cells. The scientist now adds a frozen sample of the original virus to fermenters 2 and 3. After a second week, another sample of the original virus is added to fermenter 3.
The virion count in fermenter 1 starts to fall and by the end of the first month the virus in fermenter 1 is extinct. The virion count in fermenter 2 also falls to low numbers but the virus lingers for three weeks, before becoming stable and increasing again. After this the scientist takes a sample, and finds that the number of genes in this strain has decreased to eight.
The virus in fermenter 3 grows well and is stable for at least three months. At the end of three months the scientist takes a sample, and finds that part of the genome has been duplicated and the number of genes has increased to eleven.
The scientist now sits down to write his report, which he drafts as follows:
The human virus was not at first well-adapted to monkey cells, and it experienced strong selection in all fermenters during the first week. This seems to have given strains with error-prone polymerases an advantage, because mutations increased and virion counts rose rapidly in all fermenters. The virus in fermenter 1 (where fresh virus was not added) accumulated many mutations in essential genes, and became unviable. When fresh virus with an intact polymerase was added to fermenter 2, a less error-prone strain was created by the recombination of a partially-adapted strain with the original high-fidelity strain. This evolved into a stable strain in fermenter 2 by reducing the size of its genome. Fresh virus was added twice to fermenter 3, which allowed recombination of a fully-adapted strain with the original high-fidelity strain. In the following months, the virus, now well-adapted to its host and in the absence of strong selection, acquired a rare beneficial mutation that increased the fidelity of the polymerase, allowing the virus to increase its genome size to eleven genes.
Is the scientist’s interpretation correct?
If this analysis is right, how have scientists managed to miss it for all these years?
That is a very good question, which I find very difficult to answer.
Patrick Shaw Stewart, 16 October 2022 – 3 January 2023.
It will take me months or years to write any of this up as scientific articles. Does anyone want to help me? Please email me at patrick.ss.home@gmail.com
References
Nachman, Michael W., and Susan L. Crowell. “Estimate of the mutation rate per nucleotide in humans.” Genetics 156.1 (2000): 297-304. https://doi.org/10.1093/genetics/156.1.297
Peck KM, Lauring AS. Complexities of viral mutation rates. Journal of virology. 2018 Jul 1;92(14):e01031-17. https://doi.org/10.1128/JVI.01031-17
Otto, Sarah P., and Scott L. Nuismer. “Species interactions and the evolution of sex.” Science 304.5673 (2004): 1018-1020. https://www.science.org/doi/10.1126/science.1094072
Hill, W. G.; Robertson, Alan (1966). “The effect of linkage on limits to artificial selection”. Genetical Research. 8 (3): 269–294. doi:10.1017/S0016672300010156. PMID 5980116.
Li Plant Reproduction (2018) 31:31–41. Multilayered signaling pathways for pollen tube growth and guidance. https://doi.org/10.1007/s00497-018-0324-7
Fitzsimmons, William J., et al. “A speed–fidelity trade-off determines the mutation rate and virulence of an RNA virus.” PLoS biology 16.6 (2018): e2006459. https://doi.org/10.1371/journal.pbio.2006459
Neher, R, communication on the NextStrain discussion forum: https://discussion.nextstrain.org/t/trends-in-the-prevalence-of-private-mutations/1147
Zahavi A. Mate selection—a selection for a handicap. Journal of theoretical Biology. 1975 Sep 1;53(1):205-14. https://doi.org/10.1016/0022-5193(75)90111-3
Kajii T, Ferrier A, Niikawa N, Takahara H, Ohama K, Avirachan S. Anatomic and chromosomal anomalies in 639 spontaneous abortuses. Human Genetics. 1980 Jul;55(1):87-98. https://link.springer.com/article/10.1007/BF00329132
Hamilton, William D., Robert Axelrod, and Reiko Tanese. “Sexual reproduction as an adaptation to resist parasites (a review).” Proceedings of the National Academy of Sciences 87.9 (1990): 3566-3573. https://doi.org/10.1073%2Fpnas.87.9.3566
Francis Jr, Thomas, and Alice E. Moore. “A study of the neurotropic tendency in strains of the virus of epidemic influenza.” The Journal of experimental medicine 72.6 (1940): 717. https://doi.org/10.1084%2Fjem.72.6.717
Herfst, S., Schrauwen, E. J., Linster, M., Chutinimitkul, S., de Wit, E., Munster, V. J., … & Fouchier, R. A. (2012). Airborne transmission of influenza A/H5N1 virus between ferrets. science, 336(6088), 1534-1541. https://doi.org/10.1126/science.1213362
Wang, Ning, et al. “Serological evidence of bat SARS-related coronavirus infection in humans, China.” Virologica Sinica 33.1 (2018): 104-107. https://doi.org/10.1007/s12250-018-0012-7
This Week in Virology podcast (TWiV) 615, at 28 minutes, 50 seconds. Available on YouTube.
